|
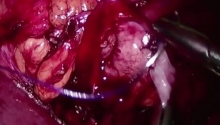
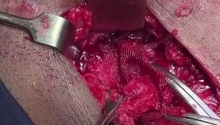
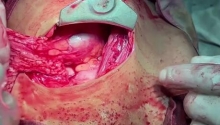
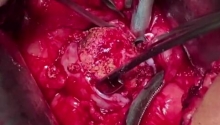
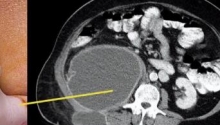
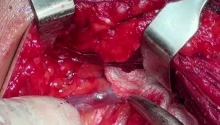
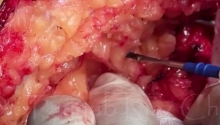
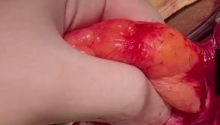
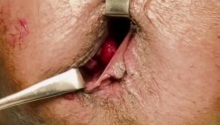
肛门闭锁是一种先天缺陷,其中没有肛门开口。这种情况发生在怀孕的第五至第七周,原因不明。它影响大约每 5,000 个新生儿中的一个,并且在男孩中比在女孩中更常见。肛门闭锁通常伴有其他先天缺陷,例如椎骨缺陷、心脏问题、气管食管瘘、肾异常和肢体异常,统称为 VACTERL 关联。这些被归类为低或高类型。在直肠靠近皮肤的低位型中,可能伴有肛门狭窄,或者肛门可能完全缺失,直肠末端为盲袋。在直肠位于骨盆上方的高位型中,直肠与膀胱、尿道或阴道之间可能存在瘘管。诊断是通过在出生后进行体检来做出的。腹部 X 光片和腹部超声有助于揭示异常的程度。治疗是通过手术创建一个开口或新的肛门,让粪便通过。手术的类型不同,取决于肛门在骨盆中的末端是高还是低。在低位型的情况下,通过一次手术打开肛门,将直肠拉下至肛门。对于高型,手术矫正分三个阶段进行。第一个程序是将肠道从腹部取出,形成造口;第二个程序是将直肠拉到肛门,在那里创建一个新的肛门;第三个程序是关闭肠造口。在这里,介绍一例 9 个月大的男性,他天生就有高型肛门闭锁。后矢状肛门直肠成形术 (PSARP) 是三个治疗阶段中的第二个。第一个是紧急乙状结肠造口术,第三个是在 PSARP 后约 6 至 8 周内关闭结肠造口术。 |

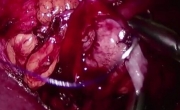 腹腔镜肾盂成形术
腹腔镜肾盂成形术
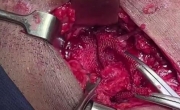 成功移除感染的腹股沟疝网片
成功移除感染的腹股沟疝网片
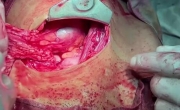 输卵管妊娠破裂的输卵管切除术
输卵管妊娠破裂的输卵管切除术
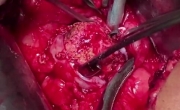 肾脏大型鹿角结石移除术_肾盂切开取石术
肾脏大型鹿角结石移除术_肾盂切开取石术

 发表于 2022-1-13 00:00:23
发表于 2022-1-13 00:00:23
 提升卡
提升卡 置顶卡
置顶卡 沉默卡
沉默卡 喧嚣卡
喧嚣卡 变色卡
变色卡 千斤顶
千斤顶 显身卡
显身卡 楼主
楼主 发表于 2022-5-19 23:21:31
发表于 2022-5-19 23:21:31